olvasási idő: 10 perc
nehézségi szint: közepes
Definíció
A sclerosis multiplex (SM) a központi idegrendszer krónikus betegsége, melynek neurológiai tüneteit az axont szigetelő myelinhüvely gyulladásos károsodása (demyelinizáció), valamint magának az axonnak, illetve az idegsejtnek a pusztulása okozza. A betegség BNO kódja G35H0, ahol a „G” az idegrendszeri érintettségre utal, azon belül pedig a központi idegrendszer demyelinizációs betegségei közé tartozik („35”), további alkategóriák nélkül („H0”).
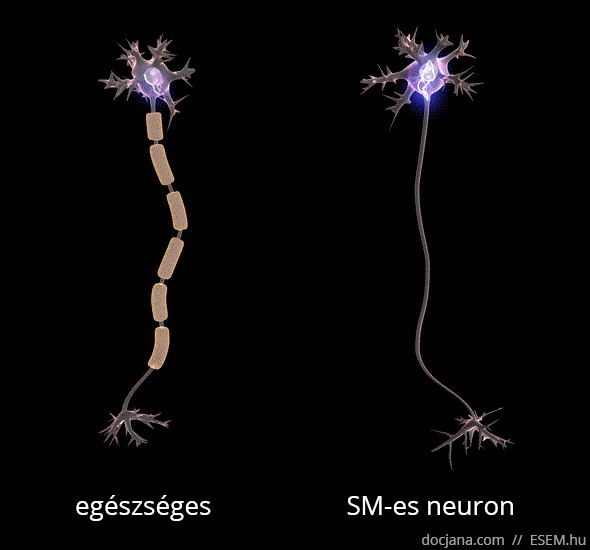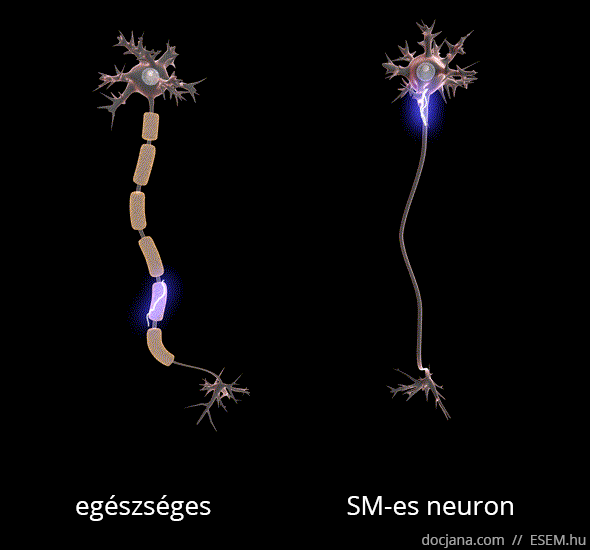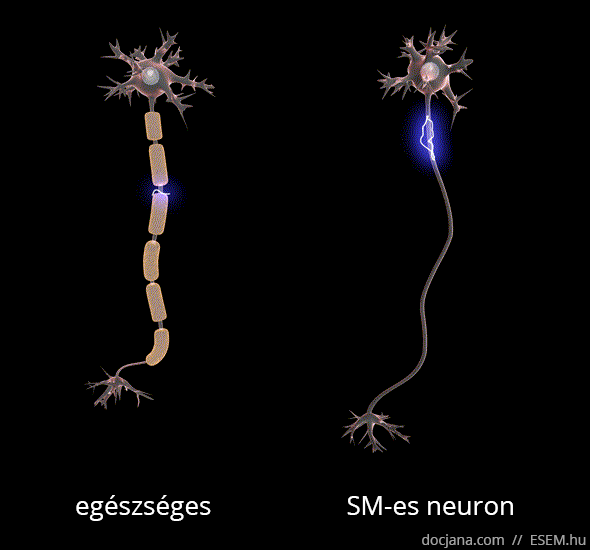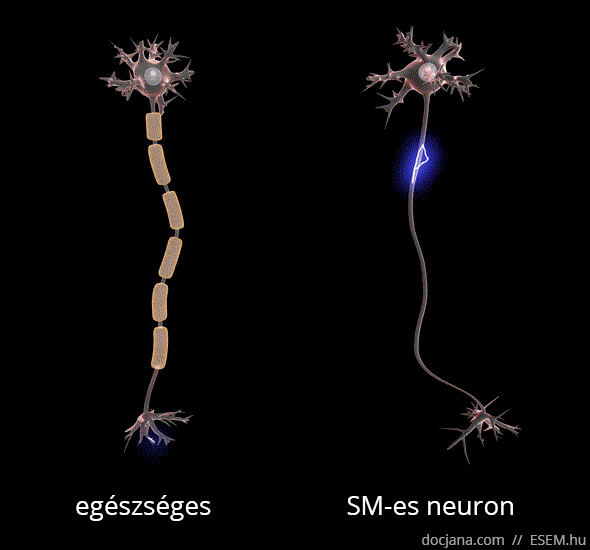
Az axon feladata az idegsejtben létrejött elektromos ingerület továbbítása a célsejt felé. A myelinhüvely az axont elektromosan szigetelő védőburok, ami az ingerület gyors és gördülékeny közvetítését segíti elő. A myelinhüvely sérülése következtében az idegsejtek vezetési sebessége lelassul, és megnyúlik az egymást követő akciós potenciálok kiváltásához szükséges idő [1], ennélfogva az idegsejtek közötti információszállítás lelassul, de fokozottabb károsodás esetén akár teljesen megszűnhet a sejtek közötti kommunikáció.
A demyelinizálódott területeken megkeményedett, elmeszesedett szövet, azaz funkcionálisan működésképtelen plakk vagy sclerosis marad. Ezek az ingerületvezetési akadályok okozzák a betegség változatos klinikai tüneteit, attól függően mást és mást, hogy a nagyagy, a kisagy, az agytörzs, a gerincvelő vagy a látóidegek mely területén keletkeztek sérülések. A betegség a nevét tehát a demyelinizáció után visszamaradt hegek többszörös jelenlétéről (multiplex) kapta [2].
A demyelinizáció mellett az SM-et az idegsejtek és kapcsolataik jól körülírt területekre korlátozott (fokális) pusztulása, azaz neurodegeneráció is kíséri, ami hosszú távon az agy kiterjedt sorvadásához, azaz agyi atrófiához vezet [3,4,5,6].
Előfordulás
Az SM a leggyakoribb neuroimmunológiai – azaz az idegrendszert fertőzés miatti vagy autoimmun eredetű immunválasz során károsító – megbetegedés, fiatal felnőttkorban pedig a mérsékelt égövön az idegrendszeri traumás sérülések és az epilepszia után a leggyakoribb neurológiai kór [7]. Előfordulása ezen a területen 60-80 fő 100 000 lakosra vetítve [4], azaz Magyarországon napjainkban 6-8000 embert érint.
A betegség jellemzően 20 és 40 éves kor között jelentkezik, 15 éves kor előtt, illetve 50 éves kor felett történő fellépése ritka [2]. Az SM két-háromszor gyakrabban fordul elő nőknél, mint férfiaknál [8]. Ennek oka egyelőre bizonytalan, de a legújabb felvetések szerint az is közrejátszhat benne, hogy egy, a tesztoszteron jelenlétében termelt molekula meggátolja a myelinhüvely pusztításáért felelős immunsejtek termelődését [9].
Kiváltó okok
Az SM egy autoimmun betegség. Az autoimmun betegségekben az immunrendszer a test saját szövetei ellen indít kóros immunválaszt. Az autoimmun eredetű betegségek kialakulása általában nem vezethető vissza egyetlen okra, hanem elsődlegesen a genetikai faktorok, továbbá a saját antigéneket módosító fertőzések, az immunrendszer működését is befolyásoló egyéb betegségek, a hormonális jellemzők és egyéb külső tényezők (pl. gyógyszerek) együttesen befolyásolják a kifejlődésüket [10].
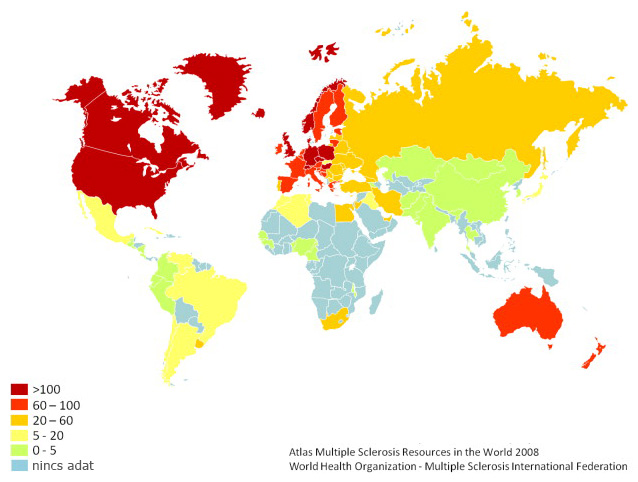
Bár az SM pontos oka ismeretlen, de kialakulásában genetikai és környezeti tényezőknek egyaránt szerepük van: „a sclerosis multiplex multifaktoriális betegség: vagyis genetikailag fogékony egyedben, egy meghatározott fogékony időperiódusban környezeti faktor(ok) vezet(nek) a betegséghez” [11 pp.51]. A genetikai tényezők bizonyítéka, hogy a betegségre való hajlam öröklődik, és már az ebben feltételezhetően szerepet játszó egyes gének is ismertek [12]. A környezeti tényezők szerepét igazolja, hogy a betegség előfordulása földrajzilag változó, méghozzá a Föld szélességi fokainak megfelelően: az Egyenlítőhöz közel az SM szinte teljesen ismeretlen, míg a legmagasabb prevalencia az északi országokra jellemző [13]. Ez a különbség az eltérő előfordulási gyakoriságú helyről fiatal korban átköltözött személyek SM-re való hajlamában is megfigyelhető [14].
Az SM megjelenési formái
Típusát tekintve az SM neurodegeneratív kórkép. Az ilyen betegségekben az anatómiai és élettani értelemben azonos rendszerhez tartozó neuronpopulációk progresszív kórlefolyással károsodnak és pusztulnak. Ez a folyamat klinikai szinten az érintett funkciók alacsonyabb szintű működését eredményezi [15].
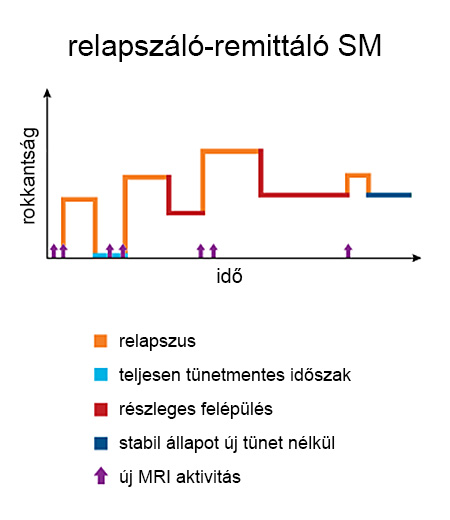
A betegség lefutása igen változó. A kezdeti fázisban a demyelinizációt a myelinhüvelyt fenntartó oligodendrocita sejtek tevékenysége még ellensúlyozza a myelinhüvely újraépítésével (remyelinizáció), és ennek köszönhetően a klinikai tünetek akár teljesen megszűnhetnek. A neuroplaszticitás és a kognitív tartalék még további kompenzációt biztosítanak. Ebben a kezdeti szakaszban a tünetekkel járó fellángolásokat (relapszusokat) a funkciócsökkenés átlagosan 1-2 hónapig tartó [3] javulása (remisszió) követi, és az egyes fellángolásokat új tünetektől mentes időszakok választják el egymástól. Ezt az időszakot relapszáló-remittáló fázisnak nevezik.
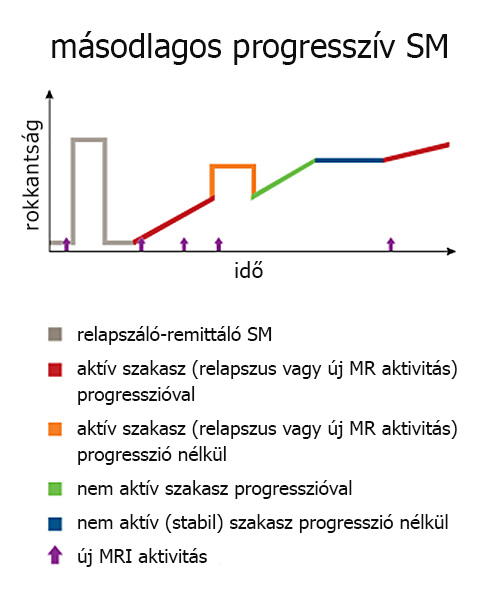
Előrehaladottabb állapotban a remyelinizáció képességének folyamatos csökkenése [5], a kognitív tartalék kimerülése [16] és a tartós axonkárosodás miatt a tünetek már nem múlnak el, így a beteg állapota fokozatosan romlik [2]. Ezt a későbbi állapotot másodlagos progresszív fázisnak nevezik. A relapszáló szakaszból a progresszív szakaszba történő átlépésre az idő előrehaladtával egyre nagyobb az esély: a kór felbukkanásakor érvényes 30% körüli esély ötévente 9%-kal növekszik, végeredményképp a betegek 65-70%-a vált át progresszív fázisra élete folyamán [17].
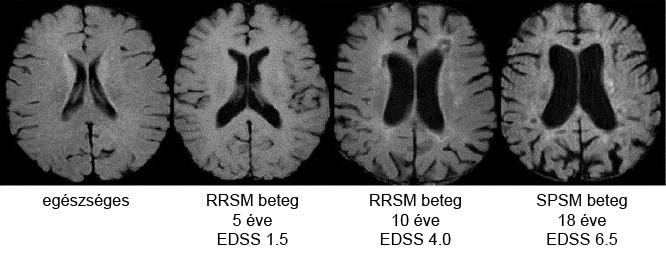 A korábbi elképzelések úgy tartották, hogy az SM főképpen a myelinhüvely és az oligodendrocita sejtek betegsége [18]. A jelenlegi álláspont szerint a betegséget a gyulladásos demyelinizáció mellett és attól függetlenül kezdettől fogva mindvégig neurodegeneráció, azaz az idegsejtek előrehaladott pusztulása kíséri [3,4,5,6]. Ez a folyamat jól megfigyelhető MRI felvételeken az agykamrák kóros tágultságának és az agytekervények patológiás kiszélesedésének formájában, valamint az agytörzs és a gerincvelő sorvadtabb állapotából [5], de szövettani elemzéssel a szabad szemmel normálisnak tűnő fehérállományban is láthatóak a jelei. A szöveti leépülés következtében az agy össztömege csökken (agysorvadás vagy agyi atrófia), és ennek mértékével a beteg klinikai állapota is szorosan korrelál [5,6]. Az agyi atrófia az öregedéssel járó természetes folyamat, de SM-ben ez legalább háromszor, súlyos esetben pedig akár tízszer gyorsabban zajlik, mint egészséges személyeknél [19]. Noha a gyulladásos demyelinizáció és az agyi atrófia párhuzamosan zajlik, a két mechanizmus egymáshoz viszonyított aránya egyénre jellemző lefutást mutat [3,5].
A korábbi elképzelések úgy tartották, hogy az SM főképpen a myelinhüvely és az oligodendrocita sejtek betegsége [18]. A jelenlegi álláspont szerint a betegséget a gyulladásos demyelinizáció mellett és attól függetlenül kezdettől fogva mindvégig neurodegeneráció, azaz az idegsejtek előrehaladott pusztulása kíséri [3,4,5,6]. Ez a folyamat jól megfigyelhető MRI felvételeken az agykamrák kóros tágultságának és az agytekervények patológiás kiszélesedésének formájában, valamint az agytörzs és a gerincvelő sorvadtabb állapotából [5], de szövettani elemzéssel a szabad szemmel normálisnak tűnő fehérállományban is láthatóak a jelei. A szöveti leépülés következtében az agy össztömege csökken (agysorvadás vagy agyi atrófia), és ennek mértékével a beteg klinikai állapota is szorosan korrelál [5,6]. Az agyi atrófia az öregedéssel járó természetes folyamat, de SM-ben ez legalább háromszor, súlyos esetben pedig akár tízszer gyorsabban zajlik, mint egészséges személyeknél [19]. Noha a gyulladásos demyelinizáció és az agyi atrófia párhuzamosan zajlik, a két mechanizmus egymáshoz viszonyított aránya egyénre jellemző lefutást mutat [3,5].
Az SM esetek 10%-ában a relapszáló szakasz teljesen kimarad [20,21], és a beteg állapota a kezdetektől fogva folyamatosan romlik, mert lefolyásában inkább a neurodegeneratív folyamatok dominálnak; ezt elsődleges progresszív típusnak nevezik.
Az egyes kórtípusokon belül az egyéni különbségek igen jelentősek. A betegség kezdetének időpontját, a kórlefolyás időtartamát és a progresszió mértékét a genetikai tényezők számottevően befolyásolják [15], ugyanakkor a környezeti faktorok epigenetikai úton is – azaz a DNS szekvenciák megváltoztatása nélkül, pusztán a génkifejeződés módosításán keresztül szabályozva a gének működését – befolyásolják a betegséglefolyást [22]. Az SM „súlyosságának” egyedi esetekre szabott előrejelzésére az orvostudománynak egyelőre nincsenek módszerei, csupán néhány irányadó megfigyelés ismert. A legelső tünet utáni felépülés elégtelensége, az első és második relapszus közötti rövid időintervallum, valamint az első tünetként jelentkező vizeletürítési vagy székelési zavarok mind rosszabb prognózist sugallnak [23].
Diagnózis
Az SM-nek nincs semmi olyan klinikai, laboratóriumi vagy radiológiai jellemzője, ami kizárólagosan csak rá illik és egyértelműen elkülöníti más betegségektől. A differenciáldiagnózis az SM jellegzetes kórlefolyása miatt a kórtörténet alapos figyelembevételével zajlik. A vizsgáló eljárások elsődleges eszköze az MRI vizsgálat, ami érzékenyen és megbízhatóan kimutatja a betegség sajátos radiológiai elváltozásait. Ezt kiegészíthetik a kiváltott válasz vizsgálatok és az agy-gerincvelői folyadék analízise. Ez utóbbinak célja az idegrendszeri gyulladás autoimmun eredetének kimutatása [18]. Ezek az eljárások azonban mindössze adalékul szolgálnak az orvos számára, támogatva vagy csökkentve az SM diagnózisának esélyét, de önmagában sem az MRI felvétel, sem a kiváltott potenciál vizsgálat, sem a liquor elemzése nem képes sem bizonyítani, sem kizárni az SM-et [3].
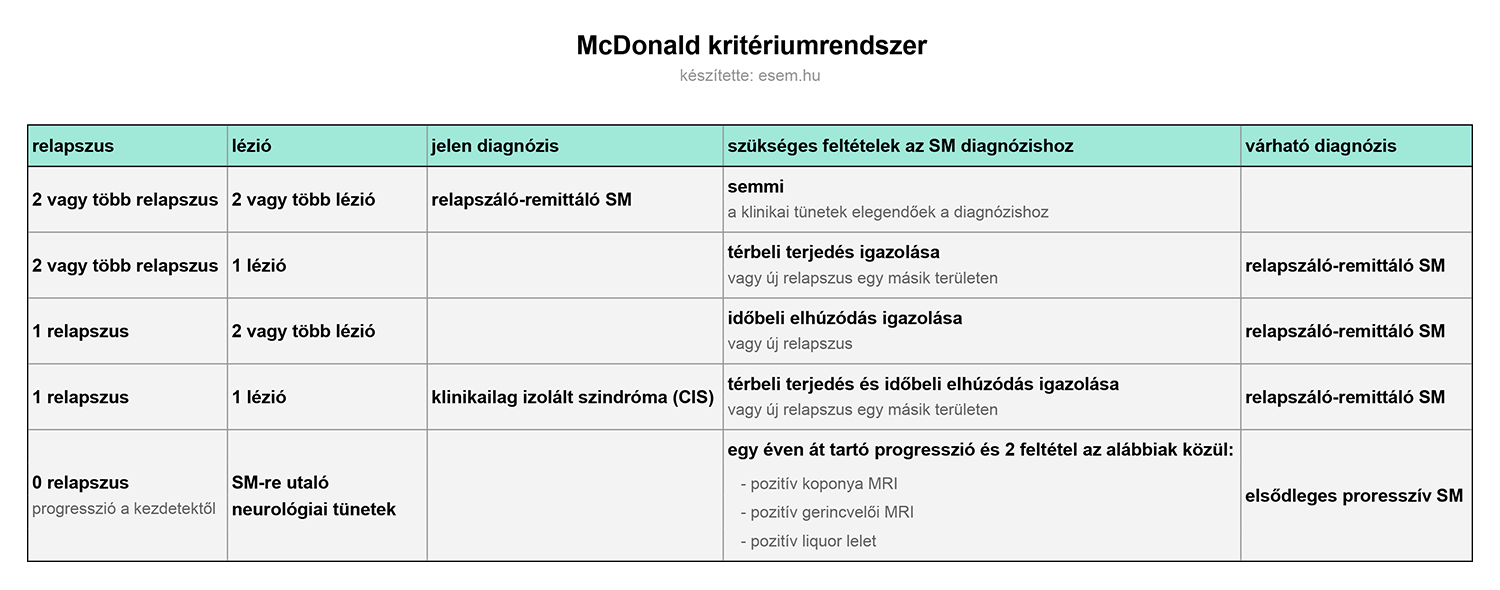
Az első tünet megjelenésekor még nem lehetséges az SM diagnózisa, az ehhez szükséges ún. McDonald-féle kritériumok ugyanis megkövetelik a betegség térbeli terjedését és időbeli elhúzódását a hivatalos diagnózis felállításához [3]. Ez azt jelenti, hogy bizonyítani kell az idegrendszer több, egymástól anatómiailag független területének érintettségét, valamint azt, hogy azokra nem egyszerre, hanem különböző időpontokban csapott le a kór. Az első tünet tehát csak az SM gyanúját vetheti fel, és mivel mindenképpen meg kell várni hozzá a következő klinikai epizódot, a diagnózis felállítása gyakran évekig elhúzódik.
Tünetek
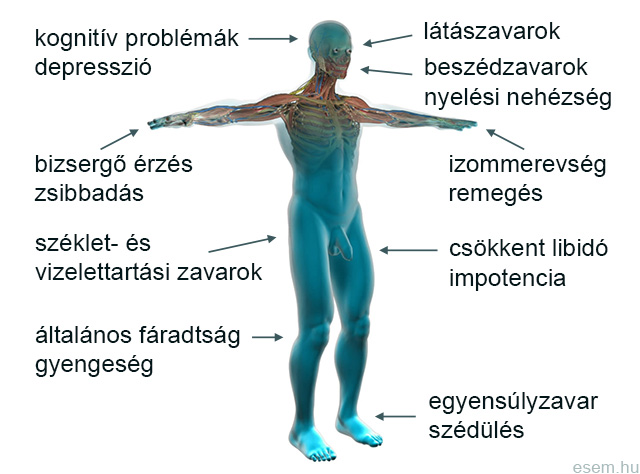
Az SM megjelenése olyan sokféle lehet, hogy aligha akad két olyan ember, akik a tünetek azonos kombinációjával küzdenek. Ezért is hívják a betegséget „ezerarcú kórnak”. A leggyakoribb tünet a patológiás fizikai és szellemi fáradékonyság (fatigue), amit valószínűleg az idegrendszerben zajló folyamatos autoimmun gyulladás okoz [2]. A fáradékonyság a páciensek háromnegyedét érinti, és a betegek 40%-a szerint ez a tünet rontja legmarkánsabban az életminőséget [24]. További gyakori tünetek a zsibbadás- vagy bizsergésszerű érzészavarok (paresthesia), a látóideggyulladás és egyéb látászavarok, a görcsös izommerevség (spasticitás), a kóros izomgyengeség (paresis), a koordinációs és egyensúlyzavarok, a vizeletürítési és székelési problémák, a szexuális diszfunkciók és a neuropátiás jellegű fájdalom [1,3,25].
Felhasznált szakirodalom
- Fuller, G., Manford, M. (2006). Neurológia. Elsevier Limited, Oxford, UK
- Komoly, S., Palkovits, M. (2011). Gyakorlati neurológia és neuroanatómia. Medicina Kiadó, Budapest
- Csépány, T., Illés, Zs. (2014). Klinikai neuroimmunológia. Matyus-Benten, Pécs
- Simó, M. (2009). A kiváltott válasz vizsgálatok jelenlegi helye a neurológiai diagnosztikában (Doktori értekezés)
- Lovas, G. (2008). A központi idegrendszeri mielint érintő megbetegedések és kezelési lehetőségeik. In: Huszti, Zs., Kálmán, M. (szerk.), Glia (507-538). Akadémiai Kiadó, Budapest
- Stankiewicz, J. M., Buckle, G. J. (2011). MS: Clinical Features, Symptom Management, and Diagnosis. In: Rizvi, S. A., Coyle, P. K. (szerk.), Clinical Neuroimmunology (89-110). Humana Press, New York, USA
- Komoly, S. (2003). Neuroimmunológiai betegségek korszerű diagnosztikája és kezelése. Magyar Tudomány, 2003(9) 1184-1192.
- Alonso, A., Hernán, M. A. (2008). Temporal trends in the incidence of multiple sclerosis: a systematic review. Neurology, 71(2), 129-35.
- Russi, A. E., Ebel, M. E., Yang, Y., Brown, M. A. (2018). Male-specific IL-33 expression regulates sex-dimorphic EAE susceptibility. Proceedings of the National Academy of Sciences, 115(7), 1520-1529.
- Gergely, J., Sármay, G. (2012). Tolerancia és autoimmunitás. In: Erdei, A., Sármay, G., Prechl, J. (szerk.), Immunológia (467-499). Medicina Kiadó, Budapest
- Illés, Zs. (2003). Sclerosis multiplex és autoimmunitás az ezredfordulón. Neurológiai Klinika, Pécs
- Gourraud, P., Harbo, H. F., Hauser, S. L., Baranzini, S. E. (2012). The genetics of multiple sclerosis: an up-to-date review. Immunological Reviews, 248(1), 87-103.
- Simpson, S., Blizzard, L., Otahal, P., Van der Mei, I., Taylor, B. (2011). Latitude is significantly associated with the prevalence of multiple sclerosis: a meta-analysis. Journal of Neurology, Neurosurgery, and Psychiatry, 82(10), 1132-1141.
- Alter, M., Kahana, E., Loewenson, R. (1978). Migration and risk of multiple sclerosis. Neurology, 28(11), 1089-1093.
- Kapás, I. (2015). Neurodegeneratív proteinopátiák klinikopatológiai elemzése. (Doktori értekezés).
- Schwartz, C. E., Quaranto, B. R., Healy, B. C., Benedict, R. H., Vollmer, T. L. (2013). Cognitive reserve and symptom experience in multiple sclerosis: a buffer to disability progression over time? Archives of Physical Medicine and Rehabilitation, 94(10), 1971-1981.
- Scalfari, A., Neuhaus, A., Daumer, M., Muraro, P. A., Ebers, G. C. (2014). Onset of secondary progressive phase and long-term evolution of multiple sclerosis. Journal of Neurology, Neurosurgery & Psychiatry, 85(1), 67-75.
- Ács, P. (2016). Immunrendszer diszfunkciójából eredő idegrendszeri elváltozások. Neurológiai autoimmun betegségek. In: Csernus, V., Kállai, J., Komoly, S. (szerk.), Emberi életfolyamatok idegi szabályozása – a neurontól a viselkedésig. Interdiszciplináris tananyag az idegrendszer felépítése, működése és klinikuma témáiban orvostanhallgatók, egészség- és élettudományi képzésben résztvevők számára Magyarországon.
- Joy, J. E., Johnston, R. B. Jr. (2001). Multiple Sclerosis: Current Status and Strategies for the Future. National Academy Press, Washington, D.C., USA
- Koch, M., Kingwell, E., Rieckmann, P., Tremlett, H. (2009). The natural history of primary progressive multiple sclerosis. Neurology, 73(23), 1996-2002.
- Koch, M., Kingwell, E., Rieckmann, P., Tremlett, H. (2010). The natural history of secondary progressive multiple sclerosis. Journal of Neurology, Neurosurgery & Psychiatry, 81(9), 1039-1043.
- LaGanke, C. (2016. május 4.). Patient Self-Advocacy Through Knowledge (Videó letöltve: 2018. 02. 25-én: https://youtu.be/h3sV7EN8mqw)
- Langer-Gould, A., Popat, R. A., Huang, S. M., Cobb, K., Fontoura, P., Gould, M. K., Nelson, L. M. (2006). Clinical and demographic predictors of long-term disability in patients with relapsing-remitting multiple sclerosis: a systematic review. Archives of Neurology, 63(12), 1686-1691.
- Charvet, L. E., Kluzer, B., Krupp, L. B. (2014). Invisible Symptoms of MS: Fatigue, Depression, and Cognition. In: Samkoff, L. M., Goodman, A. D. (szerk.), Multiple Sclerosis and CNS Inflammatory Disorders (114-121). Wiley-Blackwell, Oxford, UK
- Szirmai, I. (2011). Neurológia. Medicina Kiadó, Budapest


